









2138cn太阳集团古天乐应用物理与技术研究中心陈默涵研究员及其研究生路登辉,与北京应用物理与计算数学研究所的王涵副研究员,中国科学院计算技术研究所贾伟乐副研究员,劳伦斯伯克利国家实验室与加州大学伯克利分校的Lin Lin(林霖)教授,普林斯顿大学的Linfeng Zhang(张林峰)博士、Roberto Car教授和Weinan E(鄂维南)教授合作,研究工作“Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning”入围2020年国际高性能计算领域最高奖“戈登贝尔奖”的决赛名单(Finalist)。
分子动力学方法在物理、化学、生物及材料等科学领域有着广泛的应用。在高性能计算的快速发展下,分子动力学方法已成为阐释复杂物理化学现象的重要工具。然而,计算效率和精度不可兼得是分子动力学方法长期面临的困扰。基于经验力场的分子动力学方法效率高,但精度欠缺,而基于量子力学第一性原理(例如密度泛函理论)的分子动力学方法精度较高,但其计算量大效率低。近两年来,基于深度学习的分子动力学方法较好的结合了第一性原理方法的精度和经验力场的效率,显示出了其优势并已被快速推广使用。
在这项工作里,该研究团队在物理建模、机器学习与高性能计算的交叉学科领域取得突破,实现了深度学习分子模拟方法软件DeepMD-kit在CPU-GPU硬件架构上的编程与超大规模并行,在美国Summit超级计算机上测试了超过一亿个原子的分子动力学模拟计算,达到了双精度91 PFLOPS,混合单精度162 PFLOPS,混合半精度275 PFLOPS的峰值浮点运算能力。研究团队还采用该方法模拟了铜纳米晶粒拉伸后产生位错的过程(如图所示),这是传统第一性原理方法无法模拟的大尺度计算。
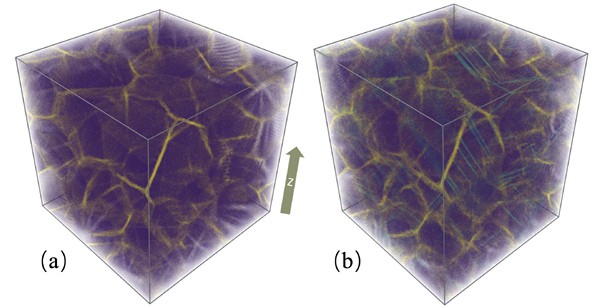
铜纳米晶粒(包含一千万个原子以上)的拉伸模拟。(a)具有面心立方晶体结构的铜原子(紫色),晶界原子(黄色)。(b)沿z轴拉伸10%后的铜纳米晶粒产生位错结构(青色)
基于深度学习的分子动力学模拟通过机器学习和大规模并行的方法,将精确的物理建模带入了更大尺度的材料模拟中,有望在将来为力学、化学、材料、生物乃至工程领域解决实际问题发挥更大作用。戈登贝尔奖的决赛结果将在11月中旬公布。